使用动态编程进行 DNA 测序
我们刚刚看到了如何找到最长的共同子序列。利用同样的原理,我们可以实现 DNA 或蛋白质测序,这对我们解决生物信息问题非常有帮助。我们将使用最流行的 Needleman-Wunsch 算法进行配准。它与我们的 LCS 算法类似,但评分系统不同。在这里,我们用不同的评分系统对匹配、不匹配和差距进行评分。该算法分为两部分:一部分是计算可能序列矩阵,第二部分是用最佳可能序列追踪实际序列。Needleman-Wunsch 算法为任何给定序列提供最佳的全局配准解决方案。由于算法本身以及评分系统的解释并不复杂,我们可以在许多网站或书籍中找到,因此我们希望将重点放在算法的实现部分。我们将把问题分为两部分。首先,我们将使用动态编程生成计算表,然后,我们将反向追踪计算表,生成实际的序列比对。在实现过程中,我们将使用 1 表示匹配,-1 表示间隙惩罚和不匹配得分。以下是我们实现的第一部分:
Unresolved include directive in modules/ROOT/pages/ch11/ch11-09.adoc - include::example$Chapter11/8.php[]在这里,我们创建了一个大小为 M,N 的二维数组,其中 M 是字符串 #1 的大小,N 是字符串 #2 的大小。我们按递减顺序将网格的第一行和第一列初始化为负值。我们将索引与间隙惩罚相乘,以实现这种行为。在这里,常数 SP 表示匹配得分点,MS 表示不匹配得分,GP 表示间隙惩罚,GC 表示间隙字符,我们将在序列打印时使用它。动态编程结束后,将生成矩阵。让我们考虑下面两个字符串:
Unresolved include directive in modules/ROOT/pages/ch11/ch11-09.adoc - include::example$Chapter11/8.php[]然后,运行 Needleman 算法后,我们的表将如下所示:
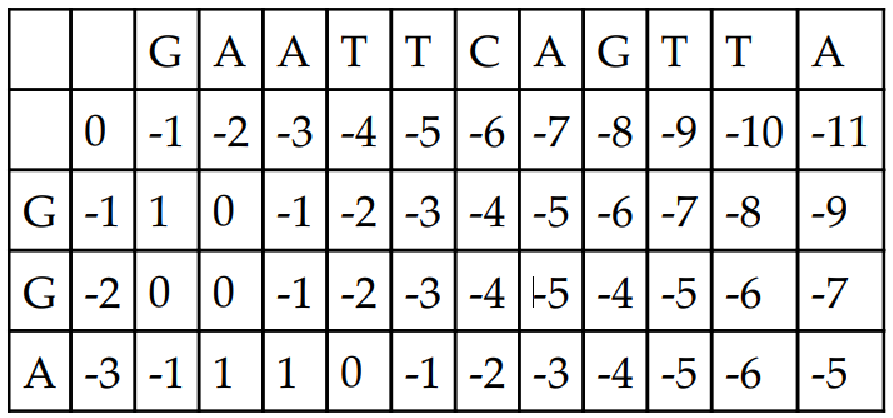
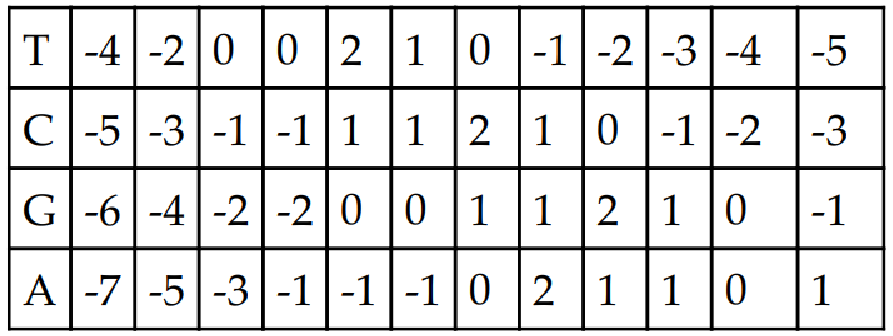
现在,我们可以利用这张评分表找出实际的序列。在此,我们将从表格中右下角的单元格开始,考虑顶部单元格、左侧单元格和对角线单元格的值。如果三个单元格中的最大值是顶部单元格,那么顶部字符串需要插入间隙字符 (-)。如果最大值是对角线上的那个,那么匹配的机会就更大。因此,我们可以比较两个字符串的两个字符,如果匹配,则可以插入一个条形或管道字符来表示对齐。下面是排序功能的外观:
Unresolved include directive in modules/ROOT/pages/ch11/ch11-09.adoc - include::example$Chapter11/8.php[]由于我们是从后面开始慢慢移动到前面的,因此我们使用数组推送来保持排列顺序。然后,我们反向打印数组。该算法的复杂度为 \$O (M * N)\$。下面是调用 NWSquencing 对两个字符串 $X 和 $Y 的输出结果:
G-AATTCAGTTA
| | | | | |
GGA-T-C-G--A